What is a marketing authorisation application?
Marketing authorisation application, or MAA, is an application that is made to a European regulatory authority for an approval to market a therapeutic drug in the European Union.
For protecting the public health and ensuring the availability of effective, high-quality, and safe medicines for European citizens, all medicines must be approved before they can be marketed.
After receiving a grant by the European Commission, the marketing authorisation (centralised) is acceptable in all EU Member States, including Iceland, Liechtenstein, and Norway.
What information is required to be submitted in an MAA?
The information submitted by the pharmaceutical companies in their application for marketing authorisation should comply with the EU legislation.
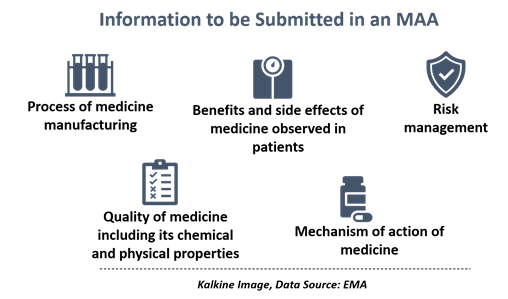
The medicine developers must include information such as the process of medicine manufacturing, its effects in laboratory studies, benefits as well as side effects observed in patients, among others.
Who is engaged in the marketing authorisation applications assessment?
A committee of experts, the CHMP, assesses the authorisation applications. Each of its members is backed by a team of assessors. The CHMP evaluates the EMA applications submitted by the manufacturer companies and advises whether a medicine should be granted marketing authorisation. During the process of evaluation, the CHMP also works with other EMA committees.
Process of obtaining an EU marketing authorisation
The European Medicines Agency (EMA) has the responsibility of conducting the scientific assessment of centralised MAA in EU. The procedure allows pharma companies to submit one MAA to the agency and to market the medicine.
Steps before submitting an application
To determine whether a product can be assessed under the centralised procedure, the applicants should always submit an eligibility request using the particular form and complemented by a justification. The steps before submitting an application are-
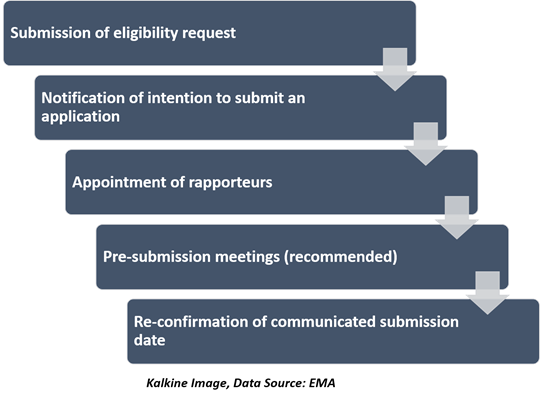
Submission of the application- After submission, the EMA performs a technical confirmation of the applications. The intention is to make sure all necessary regulatory elements needed for scientific assessment should be included in the application before commencement of the procedure.
Assessment of the application- The CHMP (Committee for Medicinal Products for Human Use) assesses MAA submitted via the centralised procedure. The evaluation is done with input from the PRAC (Pharmacovigilance Risk Assessment Committee) and the Committee for Advanced Therapies for advanced-therapy medicines. This process can take up to 210 active days.
The decision of the European Commission on the marketing authorisation- The European Commission is the authorising group for all the medicines which are authorised centrally. The Commission takes a legally binding decision based on the recommendation by the EMA.
What is the centralised procedure?
The EMA has the responsibility to evaluate the scientific evaluation of applications for centralised marketing authorisations.
After receiving grant by the European Commission, the centralised marketing authorisation is valid in all EU including Iceland, Liechtenstein, and Norway. With the grant, the marketing authorisation holder can market the medicine and provide it to patients as well as healthcare professionals all over the European Economic Area (EEA).
Which medicines require centralised authorisation?
All medicines which are for human use and derived from biotechnology or any other high-tech manufacturing methods should be evaluated by the agency through the centralised procedure.
The same is applicable for all advanced therapy medicines as well as medicinal products comprising new active substances to treat cancer, diabetes, neurodegenerative diseases, HIV/AIDS, auto-immune and other immune dysfunctions, including viral infections.
Moreover, the authorisation is also applicable for all designated orphan drugs meant to treat rare diseases.
Which medicines may optionally be centrally authorised?
For medicines which are not under any of the drugs mentioned above, the manufacturer can submit an application to the EMA, if the drug is a new active substance, has a significant therapeutic, technical, or scientific innovation.
Generics of centrally authorised products and applications for specific medicines for paediatric use may be permitted in this way. Therefore, the agency does not assess all medicines currently in use across Europe.
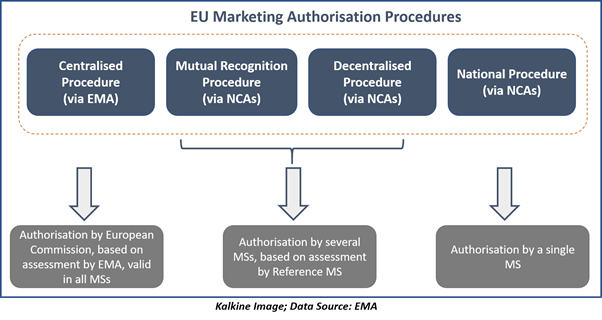
For medicines which are out of the above scope of the centralised procedure, the decentralised, mutual-recognition, or entirely national authorisation procedures should be applied, depending on the number of nations in which authorisation is required.
However, in a few some cases, when a treatment for a life-threatening disease, without any available treatment, is needed, the EMA can recommend marketing authorisation based on limited evidence on the medicine. In this case, further data is required to submit by the manufacturer at a later stage.
After a decision has been undertaken on the approval or refusal of marketing authorisation, the agency announces a complete set of documents termed as the European public assessment report (EPAR). This document comprises the public CHMP assessment report, describing the data assessed in detail and why the CHMP advised approval or rejection of authorisation.
 Please wait processing your request...
Please wait processing your request...