What are Biosimilars?
A biosimilar drug is a biological formulation developed in a way similar to an already-approved biologic, also known as the reference product. Biologics offer relatively cheaper treatment alternatives for patients suffering from chronic and often disabling indication like autoimmune disease, diabetes, and cancers.
While one might think that biosimilars are akin to generic drugs, there are significant differences between the two.
Generics are chemically identical to the branded drugs. However, a biosimilar is not a replica of another biological product. There is a level of natural variability in all biological products, and it is not possible to make an exact copy of a product that comes from living cells. All biologic, including reference products, indicate a few variations in batch-to-batch formulations.
Most of the biosimilars are likely to include biotechnology-derived proteins as an active moiety. Biosimilars are anticipated to cost from 10-40% less as compared to their reference products.
How are Biosimilars Regulated?
The EMA (European Medicines Agency) was the first body to develop an overarching structure for approving biosimilars. Biosimilars have been authorized for use in the European Union since 2006. They also have been licensed for use in Australia, Latin America, and Japan.
In the US, a regulatory pathway for approval of biosimilars was made possible by the BCPI act or Biologics Price Competition & Innovation Act. This act is a provision comprised in the Patient Protection and Affordable Care Act of 2010.
The Food and Drug Administration (FDA) gauges the safety as well as the efficacy of all the new, original biologics. Evaluation of biologics is the same as for new conventional medications. The purpose of developing a biosimilar is to determine that the proposed biosimilar is highly comparable to the reference product, with similar safety and efficacy.
What is the Approval Process for Biosimilars?
Biosimilars are approved as per the similar requirements of pharmaceutical safety, quality, and efficacy that apply to all biological medicines. In the United States, the FDA approves biosimilar products and provides the scientific and regulatory advice needed to bring safe and effective biosimilars to the market. Biosimilars can only be approved after the expiration of data exclusivity on the reference biological drug.
Biosimilar products approval could improve access to care for patients by increasing the number of alternatives for medication at potentially lower costs.
Approval by the FDA
The FDA pathway for approving reference biologics emphasizes large clinical trials that determine safety and efficacy. In contrast, the approval pathway for biosimilars places more importance on biological and physicochemical characterizations of the biosimilar molecule, because the safety & efficacy information for the reference product are readily available.
The objective of a biosimilar development program is to establish biosimilarity between the proposed biosimilar and the reference product. The program does not independently determine the safety and effectiveness of the proposed product.
Approval by the EMA
In the European Union, scientific boards of the EMA assess the majority of marketing authorization applications for biosimilar medicines earlier than they can be approved and marketed in the EU.
The European Medical Agency has issued scientific guidelines to assist developers of biosimilars confirm to the strict regulatory requirements for biosimilars approval. The guidelines also assist in providing transparency on the requirements for the approval of biosimilars.
Approval by the TGA
In Australia, biosimilars are approved by the Therapeutic Goods Administration (TGA). Some biosimilar medicines are listed on the Pharmaceutical Benefits Scheme (PBS). In August 2008, the TGA adopted several European guidelines on similar biological medicinal products.
In Australia, so far, 20 biosimilars have been approved by the TGA. Out of these 20, nine are listed under PBS and available as subsidized medicine.
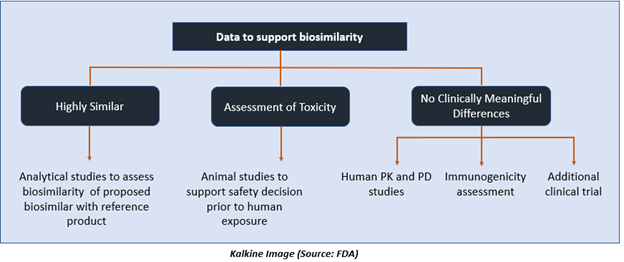
What Information is Required for the Approval of a Biosimilar?
The application for approval of biosimilar drug must include data supporting biosimilarity with the reference product. This generally comprises data from:
- Analytical studies proving that a biologic drug is highly identical to the reference drug, despite a few differences in clinically inactive components.
- Animal trials, including toxicity assessment.
- A clinical trial sufficient to establish safety, purity, as well as the potency of the proposed biosimilar product in one or more of the conditions for which the reference product is licensed.
This typically includes evaluation of immunogenicity, pharmacokinetics. In some cases, it may also include pharmacodynamics and a comparative clinical trial.
Differences between Biosimilars and Generics?
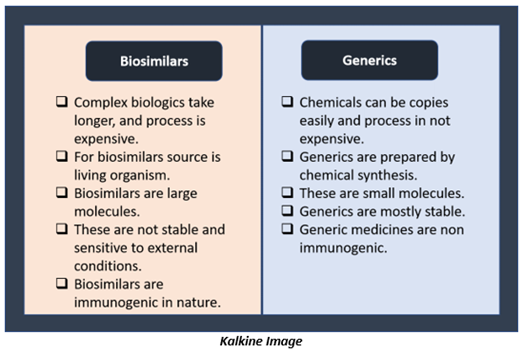
Biosimilar drugs are often confused with generic drugs. Both are marketed as lower cost as of branded drugs. These are designed to have the same clinical effect as their pricier counterparts. But biosimilars and generics are very different.
The critical difference is that generic drugs are a copy of synthetic medicines, while biosimilars are developed after drugs that use living organisms as an active ingredient.
Few key differences between biosimilar drugs and generic drugs are-
 Please wait processing your request...
Please wait processing your request...